Installing geneshot (via conda)
Written on September 22nd , 2020 by RRBIII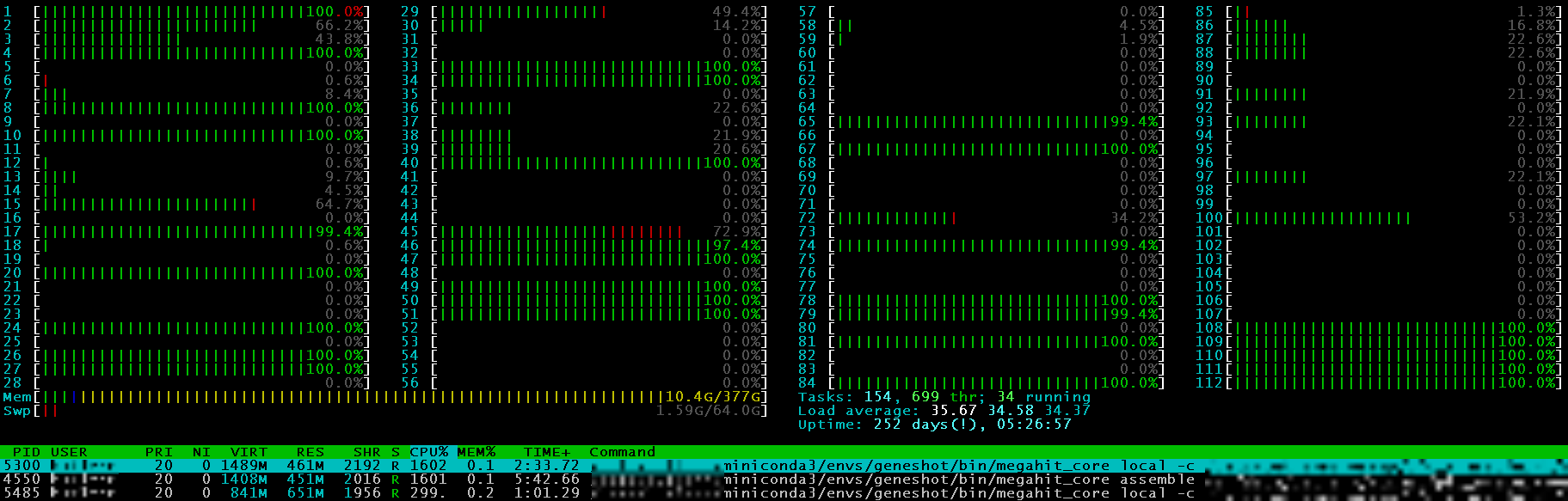
Installing geneshot (via conda)
So sometimes you can’t use docker (it happens to the best of us). In some cases, like high security environments. Geneshot is one of those neat looking new things I have been itching to try out. Luckily, miniconda is pretty flexible these days. Use an install script like:
#!/usr/bin/env bash
exec 1>> command.log 2>&1
set -ex
# build conda environment (r-breakaway built for 3.6.3)
conda create -n geneshot -y -vv python=3 nextflow r-tidyverse r-devtools \
r-vroom bioconductor-phyloseq cutadapt bwa megahit prodigal metaphlan2 diamond \
biopython mmseqs2==7.4e23d zarr pytables pyarrow
# --max-seqs option dropped in mmseqs2 >7
# pyarrow downgrades diamond quite a bit
# activate
source $(conda info --base)/etc/profile.d/conda.sh
conda activate geneshot || { echo "geneshot Conda environment not activated"; exit; }
# install non-conda components (required older python-dateutil for famli)
pip install barcodecop
pip install git+https://github.com/FredHutch/FAMLI.git@v1.5
pip install git+https://github.com/FredHutch/find-cags.git@v0.13.0
git clone https://github.com/Golob-Minot/geneshot.git -b v0.8.6
# need some fastatools to run, put them in the conda env bin folder
git clone https://github.com/Golob-Minot/fastatools.git
chmod a+x fastatools/*.py
for k in fastatools/*.py
do
j=$(basename $k)
cp $k $(conda info --base)/envs/geneshot/bin/$j
done
rm -rf fastatools
# need github R packages, for now install with R script (fix later)
#####R code
#devtools::install_github(c("adw96/breakaway", "bryandmartin/corncob"), upgrade="never")
#####
echo 'devtools::install_github(c("adw96/breakaway", "bryandmartin/corncob"), upgrade="never")' \
> devtools.R
Rscript devtools.R
rm devtools.R
Did it work?
As far as testing goes, these seem to be good to go mostly, but I get some issues running corncob. Luckily, that can be run separately with run_corncob.nf
##PASS w/o formula
NXF_VER=20.04.1 nextflow run main.nf \
-c nextflow.config \
-profile testing \
--manifest data/mock.manifest.csv \
--nopreprocess \
--output output \
--hg_index data/hg_chr_21_bwa_index.tar.gz \
--distance_threshold 0.5 \
# -w work/ \
# --noannot \
# --formula "label1 + label2" \
##FAIL
NXF_VER=20.04.1 nextflow run main.nf \
-c nextflow.config \
-profile testing \
--manifest data/mock.manifest.csv \
--preprocess_output output/preprocess_output \
--output output1 \
--hg_index data/hg_chr_21_bwa_index.tar.gz \
--formula "label1 + label2" \
--distance_threshold 0.1 \
-w work/ \
--noannot \
--savereads \
-resume
# PASS # Test with preprocessing and no formula
NXF_VER=20.04.1 nextflow run main.nf \
-c nextflow.config \
-profile testing \
--manifest data/mock.manifest.csv \
--preprocess_output output/preprocess_output \
--output output2 \
--hg_index data/hg_chr_21_bwa_index.tar.gz \
--distance_threshold 0.1 \
-w work/ \
--noannot \
-resume
# FAIL # Test with formula and no preprocessing
NXF_VER=20.04.1 nextflow run main.nf \
-c nextflow.config \
-profile testing \
--nopreprocess \
--manifest data/mock.manifest.csv \
--output output3 \
--hg_index data/hg_chr_21_bwa_index.tar.gz \
--formula "label1 + label2" \
--distance_threshold 0.1 \
-w work/ \
--noannot \
-resume
# PASS # Test with no formula and no preprocessing
NXF_VER=20.04.1 nextflow run main.nf \
-c nextflow.config \
-profile testing \
--nopreprocess \
--manifest data/mock.manifest.csv \
--output output4 \
--hg_index data/hg_chr_21_bwa_index.tar.gz \
-w work/ \
--noannot \
-resume
# PASS # Test with the gene catalog made in a previous round
NXF_VER=20.04.1 nextflow run main.nf \
-c nextflow.config \
-profile testing \
--gene_fasta data/genes.fasta.2.gz \
--nopreprocess \
--manifest data/mock.manifest.csv \
--output output5 \
--hg_index data/hg_chr_21_bwa_index.tar.gz \
-w work/ \
--noannot \
-resume
So, always good to check
Looking at the head of your data is always good, and mine runs thus far. Now to let it run through 600 samples. Even with 100 threads + 360GB memory, that will take a little bit…
# geneshot test run
head -n15 20200905_manifest.csv > test_manifest.csv
nextflow \
run \
geneshot \
--manifest test_manifest.csv \
--output test \
--nopreprocess \
-with-report \
-resume
Next time
Its worth noting, that -resume doesn’t work very well for me, it often starts over and I can’t figure out why. Also, my reads were already cleaned prior to running that, so I used --nopreprocess. However preprocessing did work in the test files, so it should be A-OK. In the future, time to solve the corncob part…