Allelic Imbalance of Expression in Rats: Part I
Written on September 21st , 2020 by RRBIII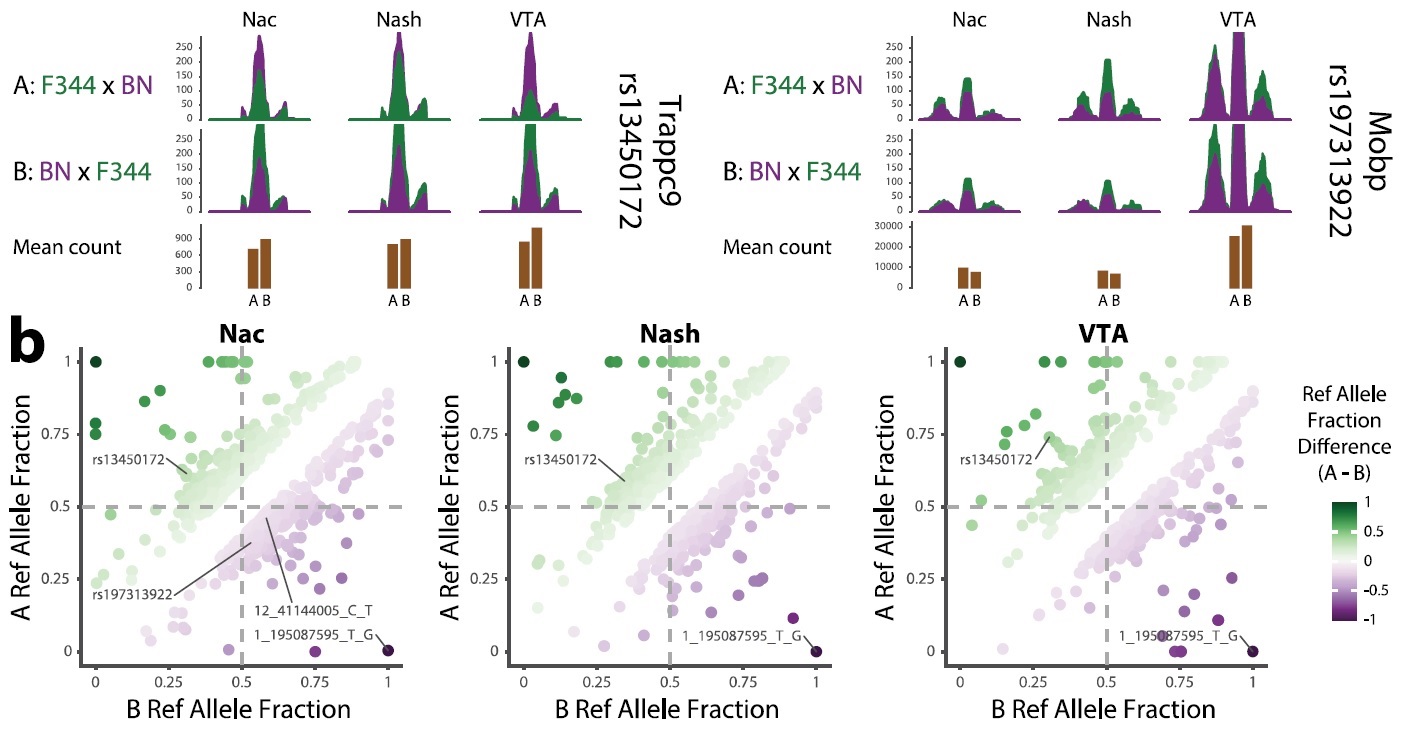
Allelic imbalance of expression part I: getting your reads
But I can’t use WASP…
Sometimes you want to look at allelic imbalance of expression and WASP isn’t an option (like when you are looking at non-human genomes, or those for which you don’t have a personal genome). The fallback method to correct for mapping bias is N-masking the SNPs in the your samples, then mapping your samples to said masked fasta [Details].
First things first, you probably already have bams from your [DE analysis]. Take a subset of your sample bams, evenly distributed across your groups and merge it into a large bam:
# list of bams is two each of three tissues for A and B (12 total)
# merge bams
samtools merge -f -@ $threads -b $bams_to_merge $merge_bam
samtools index -@ $threads $merge_bam
# merge calls
gatk --java-options "-Xmx300G" HaplotypeCaller \
-R $genome_ref \
-D $dbSNP \
--native-pair-hmm-threads $threads \
-stand-call-conf 30 \
-I $merge_bam \
-O $merge_vcf
Then you can take these merged calls and mask your genome. If you are unfamiliar with loading STAR into shared memory, check out the mapping post. Then remap:
#######################################
# masking genome
bedtools maskfasta -fi $genome_ref -bed $merge_vcf -fo $mask_fa
samtools faidx $mask_fa
#######################################
# remapping to masked fasta
# building masked STAR index
mkdir -p $STAR_INDEX
STAR \
--runMode genomeGenerate \
--runThreadN $threads \
--genomeDir $STAR_INDEX \
--genomeFastaFiles $mask_fa \
--sjdbGTFfile $GTF
# STAR-featureCounts pipeline
mkdir -p masked_bams
cd masked_bams
# add index to shared memory
STAR \
--runThreadN $threads \
--genomeDir $STAR_INDEX \
--genomeLoad LoadAndExit
# run loop
parallel -j5 --plus \
--rpl '{..} s:_R1_trim\.fastq\.gz::;s:/data/butlerr/nicotine_sensi/trimmed/::' \
STAR \
--runThreadN 20 \
--genomeDir $STAR_INDEX \
--genomeLoad LoadAndKeep \
--readFilesCommand zcat \
--outFileNamePrefix {..} \
--outSAMtype BAM SortedByCoordinate \
--outSAMmapqUnique 60 \
--outSAMattrRGline ID:{..} LB:lib1 PL:illumina PU:unit1 SM:{..} \
--outBAMsortingThreadN 10 \
--outBAMsortingBinsN 10 \
--limitBAMsortRAM 10000000000 \
--outBAMcompression 10 \
--bamRemoveDuplicatesType UniqueIdentical \
--outFilterIntronMotifs RemoveNoncanonical \
--outFilterMismatchNmax 2 \
--outFilterScoreMinOverLread 0.30 \
--outFilterMatchNminOverLread 0.30 \
--alignSoftClipAtReferenceEnds No \
--readFilesIn {} {/_R1_/_R2_} \
:::: $fasta_to_read
# remove from shared memory
STAR \
--genomeDir $STAR_INDEX \
--genomeLoad Remove
# indexing bams
parallel -j $thr_para samtools index {} ::: *.bam
cd $wk_dir
# you could do ../, but better to explicitly define your directories at the top
# of the script
Ok, now to do it all again
So, with your reads aligned with less mapping reference bias. STAR has implemented the WASP algorithm, but that is something for me to try in the future. Something that can save lots of time is gatk4.1 -ERC GVCF option. This will give you a set of gvcfs that can be combined with GenotypeGVCFs to give you an ‘all calls’ vcf with each sample called at all positions. So now you have read counts for REF and ALT everywhere (Note: I have three tissues I am looking at, so I split files up by a specific sample name pattern, then run the resulting steps in parallel -j3):
# calling gvcfs from masked bams
mkdir -p masked_gvcfs
parallel -j $thr_para --plus gatk --java-options \'"-Xmx4G"\' HaplotypeCaller \
-R $genome_ref \
-D $dbSNP \
-stand-call-conf 30 \
-I {} \
-O masked_gvcfs/{/...}${suffix2} \
-ERC GVCF \
::: $(ls masked_bams/*.bam)
cd masked_gvcfs
# tissue specific list files
ls -v *-1*Aligned_masked.g.vcf.gz | sed -r 's/(.*)Aligned/\1\t\1Aligned/' > vta.txt
ls -v *-2*Aligned_masked.g.vcf.gz | sed -r 's/(.*)Aligned/\1\t\1Aligned/' > nac.txt
ls -v *-3*Aligned_masked.g.vcf.gz | sed -r 's/(.*)Aligned/\1\t\1Aligned/' > nash.txt
# make list bed for reference genome (without patches)
awk 'BEGIN {FS="\t"}; {print $1 FS "0" FS $2}' $genome_ref.fai \
| grep '^[1-9XY]' \
> chr_list.bed
# make tissue specific gdbs (makes use of all 100 threads)
mkdir -p /data/butlerr/tmp
parallel -j3 gatk --java-options \'"-Xmx100G -Xms100G"\' GenomicsDBImport \
--genomicsdb-workspace-path {} \
--sample-name-map {}.txt \
--max-num-intervals-to-import-in-parallel 24 \
--intervals chr_list.bed \
--tmp-dir=/data/butlerr/tmp \
::: "vta" "nac" "nash"
# call Joint Genotypes
parallel -j3 gatk --java-options \'"-Xmx40G"\' GenotypeGVCFs \
-R $genome_ref \
-D $dbSNP \
-V gendb://{} \
-L $BED \
--merge-input-intervals true \
--tmp-dir=/data/butlerr/tmp \
-O {}_output.vcf.gz \
::: "vta" "nac" "nash"
One last issue though, I cannot get gatk to properly call only snps in the specified --intervals region, it just seems to ignore the option. Oh well, bcftools can get you there.
# won't filter based on --interval/-L try bcftools
parallel -j3 --plus bcftools query \
-H \
-R $BED \
-f\''%CHROM\t%POS\t%REF\t%ALT\t%ID[\t%AD\t%DP]\n'\' \
-o {..}.plot \
{} \
::: *output.vcf.gz
Next time
What to do with all that read count data.